Acute or subacute onset (days to 4 weeks)
Progressive symptoms reaching maximum within 4 weeks.
Ascending weakness (most typical feature)
Begins in legs → spreads upward to arms → facial muscles in severe cases.
Symmetric motor weakness
Weakness is usually bilateral and roughly symmetrical.
Hyporeflexia or areflexia
Deep tendon reflexes reduced or absent early.
Sensory symptoms
• Paresthesia (tingling) in feet and hands
• Mild sensory loss, but motor deficits more prominent
Autonomic involvement
• Fluctuating BP
• Postural hypotension
• Arrhythmias
• Sweating abnormalities
• Urinary retention (sometimes)
Cranial nerve involvement
• Facial weakness (bilateral facial palsy common)
• Difficulty swallowing (bulbar involvement)
Respiratory muscle weakness
May progress to respiratory failure – danger sign.
Pain
Muscle pain, radicular back pain, body aches common.
Gait difficulty
Unsteady gait due to weakness + sensory ataxia.
Variants may modify presentation
• Miller Fisher variant – ophthalmoplegia, ataxia, areflexia
• AMAN/AMSAN – more severe motor weakness, rapid decline
Respiratory failure (most serious complication)
Weakness spreads to respiratory muscles → reduced vital capacity → need for ICU and mechanical ventilation.
Occurs in ~25% of hospitalized GBS patients.
Autonomic dysfunction (common & unpredictable)
• Wide BP fluctuations (labile hypertension/hypotension)
• Cardiac arrhythmias (bradycardia, tachycardia, heart block)
• Sweating abnormalities
• Urinary retention / overflow incontinence
Can be sudden and life-threatening.
Severe pain syndromes
• Neuropathic pain (burning, shooting)
• Deep aching pain in back and legs
Pain may precede weakness.
Bulbar involvement
• Dysphagia
• Risk of aspiration pneumonia
• Facial weakness affecting airway protection
Thromboembolism risk
Immobilization → DVT and pulmonary embolism unless prophylaxis given.
Secondary infections
• Pneumonia (due to poor cough/ventilation)
• UTI (due to urinary retention or catheter use)
• Ventilator-associated infections in intubated patients
SIADH
Syndrome of inappropriate antidiuretic hormone secretion → hyponatremia.
Pressure sores
Due to prolonged immobility.
Chronic fatigue and residual weakness
Even after recovery, many patients have prolonged fatigue, distal weakness, or neuropathic symptoms.
Rare: Severe axonal variants leading to prolonged recovery
AMAN/AMSAN variants may have slow or incomplete recovery due to axonal loss.
Psychological impact
Anxiety, depression, insomnia from sudden paralysis and ICU stay.
Acute onset bilateral limb weakness
GBS causes rapidly progressive ascending weakness; other acute neuromuscular disorders can mimic it.
AIDP vs AMAN/AMSAN
Different GBS subtypes may resemble each other early and require electrophysiology for differentiation.
Acute Transverse Myelitis
• Often has a sensory level
• Early bowel/bladder involvement
• UMN signs (hyperreflexia, spasticity) — NOT typical of GBS
Spinal cord compression
• Back pain prominent
• Hyperreflexia, Babinski positive
• Emergency MRI needed if suspected
Tick paralysis
• Rapidly ascending flaccid paralysis
• Areflexia similar to GBS
• Improves dramatically after tick removal
Botulism
• Descending paralysis (opposite of GBS)
• Pupillary dilation, blurred vision, dry mouth
• No sensory symptoms
Myasthenia Gravis
• Fluctuating fatigable weakness
• Ocular, bulbar symptoms prominent
• Reflexes preserved
Lambert–Eaton Myasthenic Syndrome
• Proximal weakness
• Hyporeflexia but improves after brief exercise
• Often associated with malignancy
Acute Intermittent Porphyria
• Abdominal pain + neuropathy
• Autonomic symptoms prominent
Diphtheritic polyneuropathy
• History of pharyngeal illness
• Palatal paralysis early
• Slower progression than GBS
Toxic neuropathies
• Heavy metals
• Chemotherapy drugs
• Organophosphate poisoning
HIV-associated acute neuropathy
• May mimic GBS during acute seroconversion
• CSF pleocytosis more common
Vasculitic neuropathy
• Painful, asymmetric, multifocal
• Often mononeuritis multiplex
Critical illness polyneuropathy
• Occurs in ICU patients
• Difficulty weaning from ventilator
• No classic ascending pattern
Functional neurological disorder
• Give-way weakness
• Preserved reflexes
• Inconsistent exam findings
Immune-mediated post-infectious neuropathy
GBS is most commonly triggered by an abnormal autoimmune response following an infection.
Antecedent infections (most common triggers)
• Campylobacter jejuni — strongest association; lipooligosaccharides mimic gangliosides (GM1, GD1a).
• Cytomegalovirus (CMV).
• Epstein–Barr virus (EBV).
• Influenza virus.
• Mycoplasma pneumoniae.
• Zika virus.
• SARS-CoV-2 (COVID-19) — reported association in some outbreaks.
Vaccination-related (rare)
Historical links (e.g., 1976 influenza vaccine), but modern vaccines have extremely low risk; benefits outweigh risks.
Molecular mimicry mechanism
Pathogen antigens resemble peripheral nerve gangliosides → immune system produces cross-reactive antibodies → complement activation → demyelination and/or axonal injury.
Autoantibodies commonly detected
• Anti-GM1
• Anti-GD1a
• Anti-GT1a
• Anti-GQ1b (characteristic of Miller Fisher variant)
Subtypes influenced by etiology
• AIDP (Acute inflammatory demyelinating polyneuropathy) — most common in Europe/Asia; T-cell + macrophage-mediated demyelination.
• AMAN (Acute motor axonal neuropathy) — strongly associated with **C. jejuni**; anti-GM1 / GD1a antibodies.
• AMSAN (Acute motor-sensory axonal neuropathy) — more severe axonal variant; often infection-related.
• Miller Fisher Syndrome (MFS) — linked to **anti-GQ1b antibodies**; ophthalmoplegia.
Risk factors
• Recent gastrointestinal or respiratory infection
• Viral outbreaks (e.g., Zika-related surges)
• Older age increases severity
• Slight male predominance
Not genetically inherited
GBS is generally sporadic; no familial pattern.
Electrodiagnostic studies (cornerstone test)
Nerve conduction study (NCS) shows demyelination: slowed conduction velocity, prolonged distal latencies, conduction block. Helps differentiate subtypes (AIDP vs AMAN/AMSAN).*
Cerebrospinal fluid (CSF) – albuminocytologic dissociation
High protein with normal/low WBC count (<10 cells/mm³). Usually seen after 1 week from onset.
Anti-ganglioside antibodies
Anti-GM1, GD1a (AMAN); anti-GQ1b (Miller Fisher syndrome). Supportive but not mandatory.
Blood tests (rule out mimics)
CBC, ESR/CRP, electrolytes, HbA1c, LFT/RFT, B12, thyroid profile to exclude metabolic or inflammatory neuropathies.
Infection workup when history suggests
Stool PCR/serology for Campylobacter jejuni, viral serology (CMV, EBV, HIV, Zika), respiratory PCR if recent illness.
Respiratory monitoring
Serial FVC, NIF (negative inspiratory force). Detects early respiratory compromise.
Autonomic monitoring
Continuous BP and ECG monitoring to detect arrhythmias, BP fluctuations.
MRI spine (if needed)
May show enhancement of spinal nerve roots, cauda equina. Used mainly to rule out spinal cord compression.
NCS patterns useful for subtype identification
AIDP → demyelinating features.
AMAN/AMSAN → reduced CMAP amplitudes with minimal demyelination.
MFS → normal limbs but absent H-reflex ± abnormal oculomotor nerves.
Characteristic demyelinating features (AIDP)
• Markedly slowed motor nerve conduction velocity
• Prolonged distal motor latency
• Temporal dispersion of CMAPs
• Partial conduction block (drop in CMAP amplitude between proximal vs distal stimulation)
• Prolonged or absent F-waves (very typical early change)
• Reduced CMAP amplitudes if secondary axonal loss begins
• Sensory nerve action potentials (SNAPs) often preserved early
• Absent H-reflexes in most patients
Axonal variants (AMAN / AMSAN)
• Normal conduction velocity and latency
• Severely reduced CMAP amplitudes early
• Normal sensory responses in AMAN
• Reduced SNAPs in AMSAN
• No conduction block (distinguishes from AIDP)
• F-waves often absent due to axonal involvement
Early changes (first few days)
• F-wave absence is often the earliest abnormality
• H-reflex absent before other abnormalities appear
• Mild slowing or normal NCS early → repeat after 7–14 days improves diagnostic clarity
Typical electrophysiologic pattern in AIDP
• Demyelinating features seen in ≥2 nerves
• Prolonged F-wave latency > 120% of upper limit
• Motor conduction velocity < 70% of lower limit
• Distal latency > 130% of upper limit
• CMAP amplitude > 20% drop on proximal stimulation → conduction block
Miller–Fisher Syndrome (MFS)
• Often normal motor conduction studies
• Reduced or absent sensory nerve action potentials
• Absent H-reflex is highly sensitive
• EMG usually normal
When to repeat NCS/EMG
• If initial test is normal but clinical suspicion is high
• Best diagnostic yield between 7–14 days from symptom onset
• Helps distinguish CIDP evolution from GBS if symptoms progress beyond 8 weeks
Immune-mediated attack on peripheral nerves
GBS is caused by an abnormal immune response that mistakenly targets components of the peripheral nervous system.
Molecular mimicry is the core mechanism
Antigens of preceding infections (e.g., C. jejuni, CMV, EBV, Zika, influenza, Mycoplasma) resemble gangliosides on peripheral nerves → antibodies cross-react → immune attack.
Complement activation → myelin injury
Cross-reactive antibodies bind to gangliosides on Schwann cells or axolemma → activate complement → membrane attack complex (MAC) formation → segmental demyelination and/or axonal damage.
Macrophage-mediated demyelination
Activated macrophages penetrate the basal lamina of Schwann cells → strip myelin in a patchy, segmental pattern → slowed or blocked nerve conduction.
Node of Ranvier dysfunction
Inflammation disrupts Na⁺ channel clusters → conduction failure → acute flaccid paralysis.
Subtypes reflect the target of immune attack
• **AIDP** – autoimmune T-cell & macrophage–mediated demyelination.
• **AMAN** – antibody-mediated attack on axolemma (GM1, GD1a) → pure motor axonal injury.
• **AMSAN** – similar to AMAN but sensory axons also affected.
• **Miller–Fisher Syndrome** – anti-GQ1b antibodies → affects cranial nerves → ophthalmoplegia.
Breakdown of blood–nerve barrier
Inflammation increases permeability, allowing immune cells & antibodies to reach nerve roots and peripheral nerves.
Resulting pathophysiology
• Conduction block
• Slowed nerve conduction velocity
• Muscle weakness and areflexia
• Autonomic dysfunction in severe cases
No central nervous system involvement
GBS affects peripheral roots and nerves; the brain and spinal cord remain structurally normal.
Primary lesion
Immune-mediated demyelination of peripheral nerves and nerve roots with secondary axonal damage.
Classic CIDP pathology
Segmental demyelination, onion-bulb formations from Schwann-cell proliferation, macrophage-mediated myelin stripping.
Paranodal CIDP pathology
IgG4 autoantibodies disrupt axo-glial junctions → detachment of paranodal loops → conduction block without inflammation.
Inflammatory infiltrates
Endoneurial and perivascular T-cell and macrophage infiltration in classic CIDP.
Complement activation
Present in classic CIDP, but absent in IgG4-mediated forms.
Nerve root hypertrophy
Seen in chronic disease due to repeated cycles of demyelination and remyelination.
Secondary axonal loss
Occurs late and determines long-term disability.
No specific method to fully prevent CIDP
CIDP is autoimmune and idiopathic in most patients; no vaccine or direct preventive measure exists.
Prevent secondary worsening
Early recognition of relapse, prompt treatment, and regular follow-up reduce long-term disability.
Prevent treatment-related complications
Monitor blood sugar with steroids, avoid infections during immunosuppressive therapy, screen before rituximab.
Control modifiable triggers
Good infection control, managing chronic diseases (diabetes, HIV, hepatitis), and avoiding neurotoxic drugs if possible.
Prevent disability
Physiotherapy, gait training, orthotic support, fall-prevention, and muscle-strength preservation.
Prevent severe relapse
Patients with known autoantibody-positive paranodal CIDP require closer monitoring as relapses may be more sudden.
Patient education
Teach early danger signs—new limb weakness, falls, breathing difficulty, loss of bladder/bowel control.
Why does CIDP last longer than GBS?
GBS is acute immune activation; CIDP involves ongoing or relapsing immune activity targeting myelin.
Why is IgG4 CIDP different?
IgG4 does not activate complement → little inflammation → poor response to IVIG/steroids but good response to rituximab.
Why do some patients develop severe ataxia?
Large-fiber sensory demyelination causes proprioceptive loss → sensory ataxia.
Why do reflexes disappear early?
Demyelination slows conduction across reflex arcs → hyporeflexia/areflexia even before strength loss is severe.
Why is early treatment critical?
Delayed treatment → irreversible axonal loss → permanent disability.
Why can CIDP mimic motor neuron disease?
Motor-predominant CIDP with wasting may resemble ALS but conduction studies reveal demyelination.
Why do some patients relapse after stopping therapy?
Autoimmune activity persists; maintenance therapy is often needed.
Why is CSF protein high without cells?
Albuminocytologic dissociation → breakdown of blood-nerve barrier increases protein leakage but no inflammation in CSF.
Why nerve biopsy is rarely needed?
NCS and clinical criteria are usually sufficient; biopsy reserved for unclear or atypical presentations.
Classic CIDP
Symmetric sensorimotor weakness progressing over ≥8 weeks; good response to IVIG or steroids.
AIDP-like chronic course
Slower onset version mimicking acute GBS but evolving chronically.
Distal Acquired Demyelinating Symmetric Neuropathy (DADS)
Mainly distal sensory loss; often associated with IgM anti-MAG antibodies.
Multifocal Acquired Demyelinating Sensory and Motor Neuropathy (MADSAM / Lewis-Sumner)
Asymmetric involvement; affects individual nerves; may mimic mononeuritis multiplex.
Motor-predominant CIDP
Weakness > sensory symptoms; may resemble motor neuropathies.
Sensory-predominant CIDP
Profound sensory ataxia with minimal weakness.
Autoantibody-positive nodal/paranodal CIDP
IgG4 antibodies against NF155, NF186, CNTN1, CASPR1; poor response to IVIG but respond to rituximab.
Chronic immune sensory polyradiculopathy (CISP)
Pure sensory CIDP variant with normal nerve conduction but abnormal somatosensory evoked potentials.
Relapsing-remitting CIDP
Periods of relapse and partial recovery; requires maintenance therapy.
Progressive CIDP
Slow continuous decline without clear relapse pattern.
Symmetric flaccid weakness
Bilateral, relatively symmetric weakness—starting in legs → ascending pattern.
Hyporeflexia or areflexia
Deep tendon reflexes markedly reduced or absent in involved limbs.
Ascending motor deficit
Weakness usually begins distally in legs → spreads proximally → may involve arms, facial, bulbar, and respiratory muscles.
Cranial nerve involvement
• Facial diplegia (bilateral facial weakness)
• Dysphagia
• Dysarthria
• Ophthalmoplegia (especially in Miller–Fisher variant)
Sensory signs (mild compared to motor)
• Decreased vibration sense
• Distal paresthesias
• Reduced proprioception in some cases
Autonomic dysfunction
• Tachycardia or bradycardia
• Labile blood pressure
• Arrhythmias
• Urinary retention
• Ileus or constipation
Respiratory compromise
Weak diaphragm + accessory muscles → ↓ vital capacity → impending respiratory failure.
Gait difficulty
Steppage gait due to foot drop; inability to walk independently in progressive stages.
Pain on palpation
Paraspinal or proximal limb tenderness (due to nerve root inflammation).
Ataxia in MFS variant
Sensory ataxia with preserved motor strength → unsteady gait.
Guillain–Barré syndrome (GBS) is an acute immune-mediated demyelinating polyneuropathy.
It typically presents with rapidly progressive, symmetrical weakness beginning in the legs and ascending upward, often accompanied by sensory symptoms and areflexia.
GBS is classically a **post-infectious autoimmune disorder**, where the immune system mistakenly targets peripheral nerve myelin or axons through molecular mimicry.
Most patients develop symptoms **1–3 weeks after a preceding infection** such as Campylobacter jejuni, CMV, EBV, Mycoplasma, influenza, or other respiratory/GI pathogens.
It is the most common cause of acute flaccid paralysis worldwide.
The disease progresses for up to 4 weeks, followed by a plateau phase and gradual recovery.
Timely diagnosis and management are essential because **respiratory muscle involvement** can lead to respiratory failure requiring ventilatory support.
Although potentially life-threatening, **the majority of patients recover**, especially with early IVIG or plasmapheresis.
References
PDF
wiki
Dr Sankaran • 2025-11-27 23:48:47

Georges Charles Guillain
Dr Sankaran • 2025-11-27 23:48:24
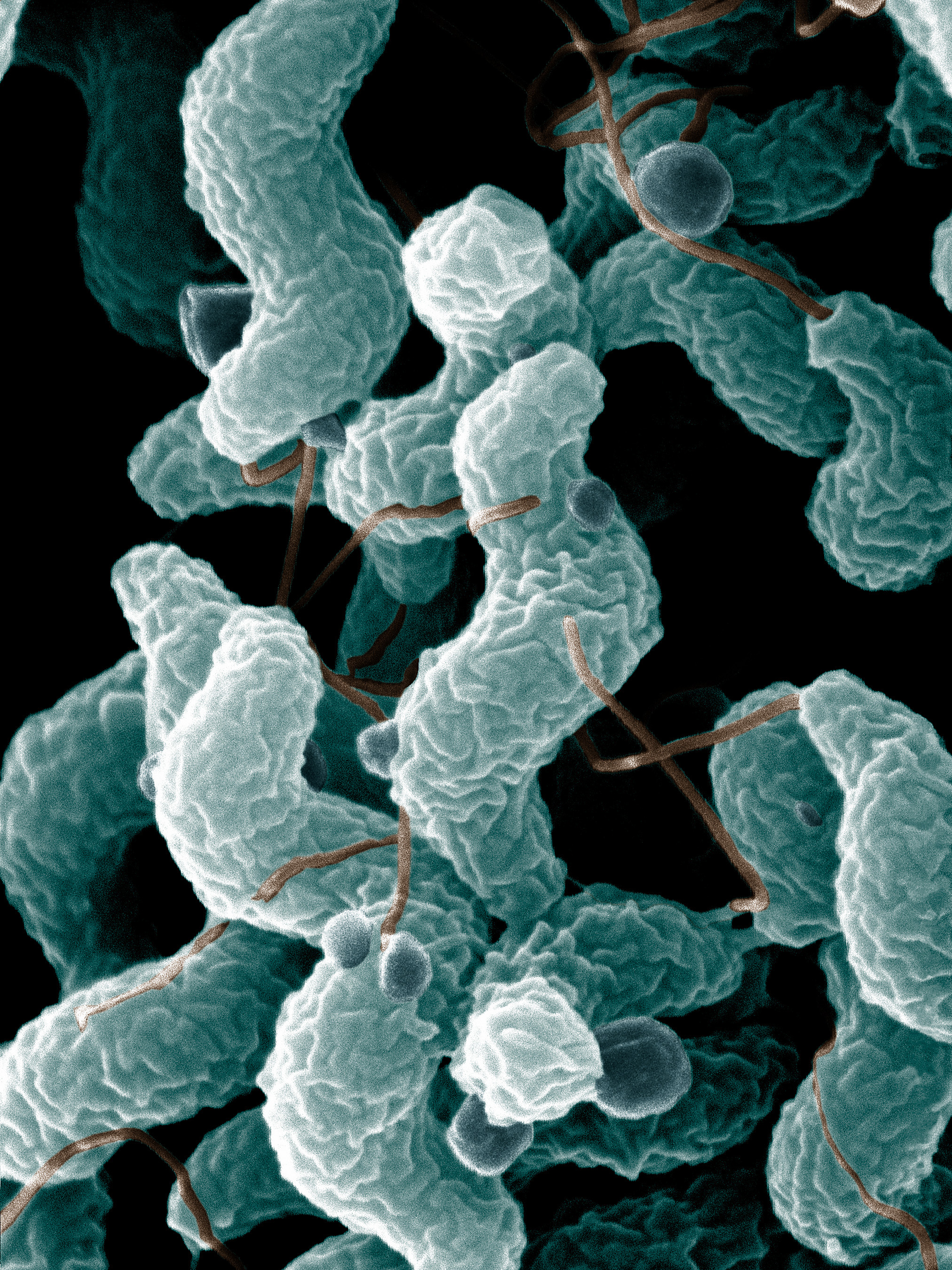
Campylobacter jejuni
Dr Sankaran • 2025-11-27 23:46:56
Progressive, symmetrical limb weakness
Weakness typically begins in the legs → ascends upwards over days to weeks.
Paresthesias
Tingling, numbness, “pins and needles” in feet and hands; often the earliest symptom.
Difficulty walking
Unsteady gait, frequent falls, inability to climb stairs or rise from sitting.
Back pain
Deep aching pain in the lower back or thighs; common early sign.
Cranial nerve involvement
• Facial weakness (bilateral facial palsy).
• Difficulty swallowing.
• Diplopia.
• Dysarthria.
Respiratory symptoms
Shortness of breath, weak cough, orthopnea — due to diaphragmatic weakness.
Autonomic symptoms
• Palpitations
• Postural dizziness
• Sweating abnormalities
• Urinary retention
• Constipation
Sensory symptoms are mild
Pain, paresthesias > objective sensory loss.
Rapid progression
Symptoms worsen over 2–4 weeks (unlike CIDP which progresses >8 weeks).
Variants may show different patterns
• MFS – ophthalmoplegia, ataxia, areflexia.
• AMAN – pure motor weakness without sensory loss.
Medical emergency – requires hospital admission
All suspected GBS patients must be monitored in a hospital (preferably ICU/HDU) due to risk of respiratory failure and autonomic instability.
When to start disease-modifying therapy
Start immediately if:
• Unable to walk independently for ≥10 meters
• Rapid progression
• Significant bulbar or respiratory involvement
• Rapidly worsening autonomic instability
First-line treatments
• Intravenous immunoglobulin (IVIG) 0.4 g/kg/day × 5 days
• Plasma exchange (PLEX) 4–6 exchanges over 7–10 days
Both are equally effective — **do NOT combine** (no added benefit).
When to choose IVIG
• Hemodynamic instability
• Elderly or frail
• Easier administration
• Poor venous access
• Slightly better in children
When to choose Plasma Exchange
• Very rapid progression
• High antibody burden (e.g., axonal variants)
• When IVIG contraindicated (renal failure, IgA deficiency)
Respiratory management
• Serial vital capacity: intubate if VC < 15 mL/kg
• Watch for: weak cough, difficulty clearing secretions
• Early ICU transfer for impending failure
Autonomic dysfunction management
• Treat arrhythmias, BP swings, urinary retention
• Avoid drugs causing bradycardia or hypotension
• Cardiac monitoring in moderate–severe cases
Pain control
• Neuropathic pain common
• Use: gabapentin, pregabalin, carbamazepine, tramadol
• Avoid heavy opioids if possible
DVT prophylaxis
• Enoxaparin or mechanical prophylaxis due to immobility
Nutritional + supportive care
• Physiotherapy as soon as medically safe
• Prevent contractures and pressure ulcers
• Speech therapy for bulbar involvement
• Early nutritional support (NG/PEG if needed)
What NOT to do
• Do NOT give corticosteroids (ineffective in GBS)
• Do NOT combine IVIG + PLEX
• Do NOT delay treatment waiting for EMG/LP reports
Monitoring
• FVC every 4–6 hours in early phase
• Continuous cardiac monitoring
• Pain and autonomic symptoms daily
• Electrophysiology if diagnosis uncertain
Expected recovery
• Majority improve within weeks
• Recovery may take months
• Axonal forms (AMAN/AMSAN) have slower recovery
Relapse / Treatment-related fluctuation
• 8–10% worsen again after initial improvement
• May require repeat IVIG course or PLEX
Long-term rehabilitation
• Strength recovery exercises
• Occupational therapy
• Assistive devices for walking
• Psychological support due to prolonged recovery period
Tap a card to view full section
Use the coloured cards above (Etiology, Symptoms, Treatment, etc.).